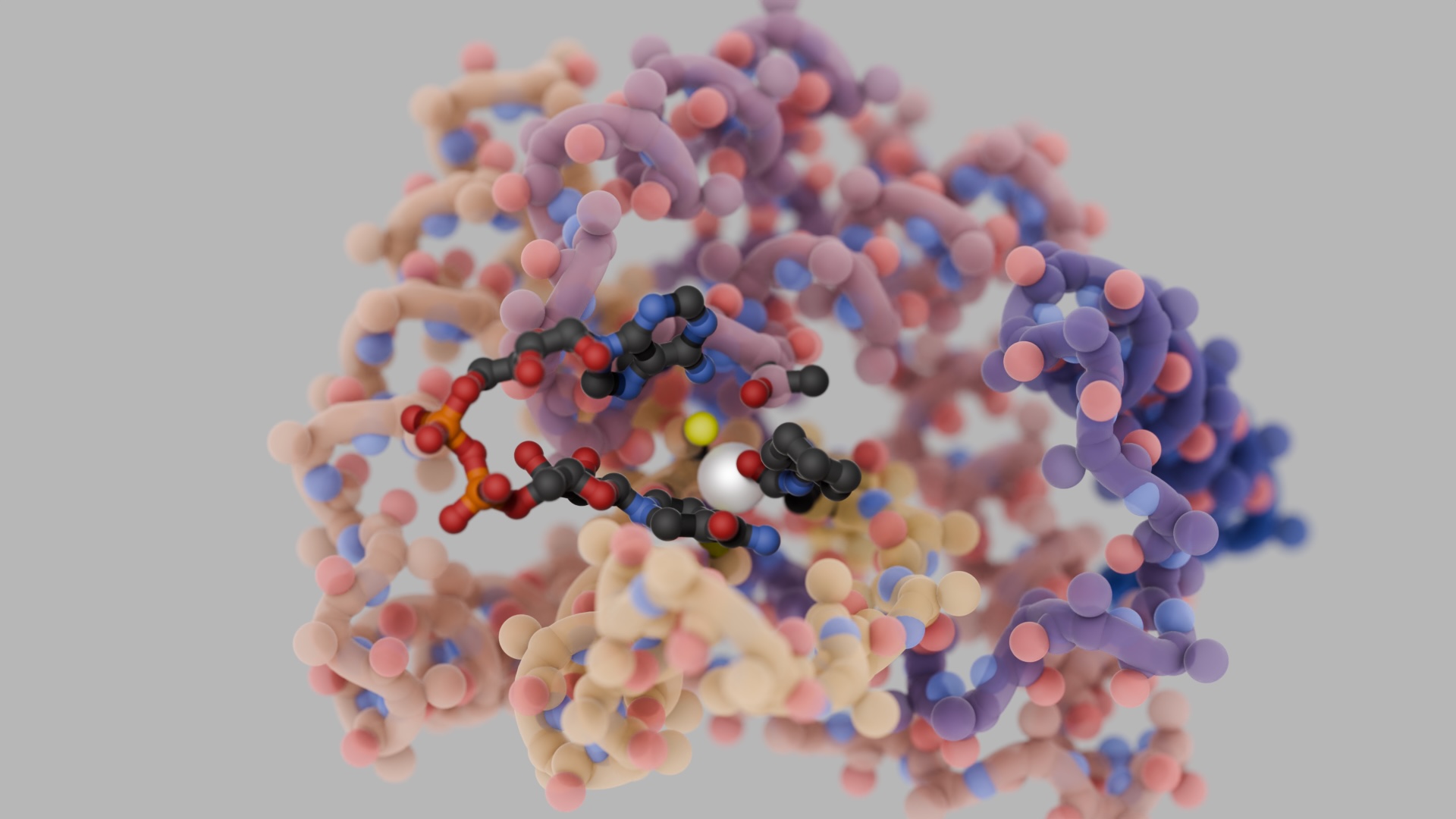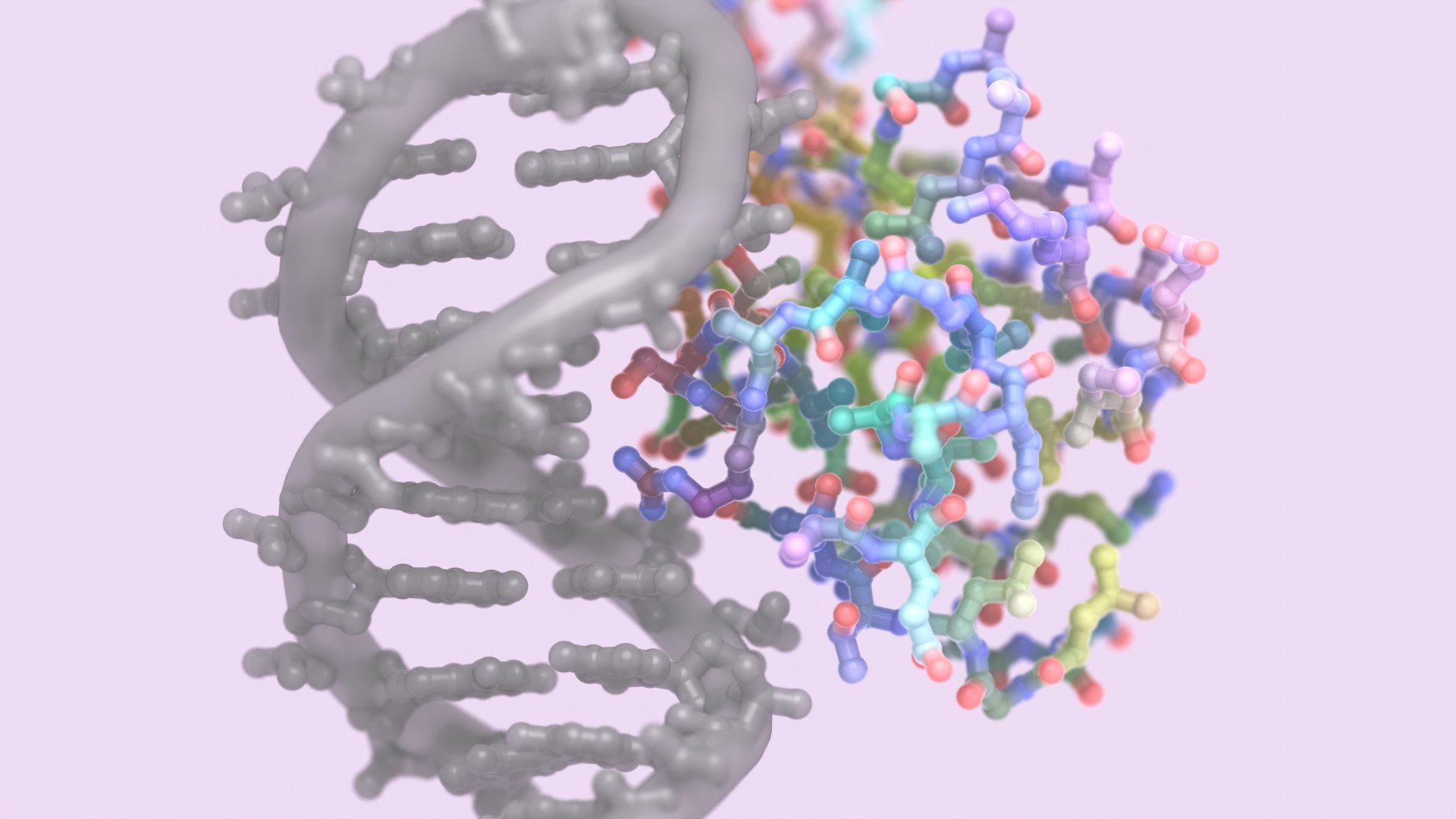
Our latest deep-learning model, LigandMPNN, excels at creating proteins that interact with other diverse molecules, including small molecules, nucleic acids, and metals. We anticipate a wave of new innovations, from highly selective biosensors for diagnostics and environmental monitoring to robust scaffolds for drug development and therapeutic delivery.
“We’re incredibly excited to see new functional proteins coming from the open-sourced LigandMPNN model, which allows designers to specify atom-level constraints for sequence design problems,” said project lead Justas Dauparas, a recent postdoctoral scholar in the Baker Lab.
Atomic context-conditioned protein sequence design using LigandMPNN
Published in: Nature Methods [PDF]
Authors: Justas Dauparas, Gyu Rie Lee, Robert Pecoraro, Linna An, Ivan Anishchenko, Cameron Glasscock, David Baker
What is sequence design?
The computational design of new proteins typically proceeds in steps. First, tools such as RFdiffusion are used to generate plausible protein architectures. This defines a molecule’s size and shape but not its full atomic details, such as amino acid side chains. To complete the protein, sequence design tools assign specific amino acid identities to each segment of the protein, yielding a complete amino acid sequence that encodes the generated structure.
Traditional sequence design focuses on optimizing side chains to stabilize the protein fold, leaving out potential interactions with atoms beyond the protein itself. But to create proteins that bind specific drugs, for example, software must properly “see” these non-protein components. Although physically based methods like Rosetta have long supported the design of such protein–ligand interactions, setting them up frequently requires custom parameterization and carefully tuned energy functions. More modern deep-learning-based sequence design tools such as ProteinMPNN have proven excellent at sequence design on protein backbones alone, but until now have not directly modeled the presence of ligands.
Enter LigandMPNN
LigandMPNN builds on ProteinMPNN to explicitly incorporate the spatial and chemical context of non-protein atoms, enabling more accurate and faster design of protein–ligand interfaces during sequence design.
In addition to the distances and angles between residues, it encodes:
- Nearest-Neighbor Ligand Atoms: By incorporating a ligand graph alongside the protein graph, LigandMPNN can learn how residues and ligand atoms interact at close range.
- Element Types: Unlike backbone-centric models, LigandMPNN recognizes chemical element identities, which is critical for binding metals and large or unusual ligands.
- Sidechain Packing: The method also predicts side-chain conformations, letting designers see how side chains might orient around target ligands.
Model Performance
These innovations together produce big improvements. When tasked with re-installing key side chains into proteins known to bind ligands, LigandMPNN achieves a 10–40% jump in sequence recovery performance compared to other approaches.
LigandMPNN has also been used extensively at the IPD, leading to over 100 new experimentally validated small-molecule and DNA-binding proteins with high affinity and high structural accuracy. This includes:
- Redesigned proteins with 100-fold improvements in binding to target compounds. In several instances, LigandMPNN successfully rescued failed small-molecule binders, turning non-binders into measurable nanomolar- to micromolar-affinity proteins.
- Designs that specifically target DNA base interactions achieved the high accuracy required for sequence-specific recognition. One of these designs was solved by X-ray crystallography, confirming the predicted binding pose.
- Installing metal-binding sites into proteins can be tricky given strict geometric constraints, but LigandMPNN facilitates these interactions in lab-tested proteins, underscoring the method’s precision.
Why This Matters
With LigandMPNN, protein sequences can now be created more accurately and hundreds of times faster than with traditional tools, enabling larger-scale protein design campaigns and iterative workflows. This tool does not require extensive hand-parameterization for each new ligand or metal, and consistent improvements in sidechain and sequence recovery around diverse types of ligand atoms points to a more robust and broadly applicable design approach.
From creating enzymes that catalyze new reactions to developing sensors that detect chemical pollutants, the ability to account for non-protein molecules while developing custom proteins is essential for next-generation applications of protein design.
Getting Started
We’ve released the open-source code for LigandMPNN so anyone can try it out in their own protein design projects. Visit the LigandMPNN GitHub repository to get started. We provide instructions on installing dependencies, preparing input PDB files with ligands or metals, and generating designed protein sequences. Example workflows show how LigandMPNN can be used to redesign a binding pocket for improved ligand affinity or specificity.
We look forward to the community’s feedback and to collaborations that will push the boundaries of what’s possible with protein design.